第一性原理计算和固体能带理论在拓扑材料的预言方面发挥了重要的作用。经过十多年的发展,基于对称性表示的拓扑能带理论也取得了重要进展,包括对称性指标理论(symmetry indicators)和拓扑量子化学理论(topological quantum chemistry),它们的理论基础都是晶体中的布洛赫电子态可以由空间群的不可约表示来标记。其中拓扑量子化学的基本思想是利用完备的局域轨道能带表示来有效地判断能带拓扑:即一个能带结构如果不是(基本)能带表示的和,那它就是拓扑的。拓扑量子化学本质上就是一套系统的空间群的表示理论,它通过分解倒空间电子态的能带表示,可以直接定位出电子在实空间的轨道特征,由此建立了“不可约表示”(物理概念)和“电子轨道”(化学概念)之间的直接联系。前期人们已经通过这些基于对称性的拓扑能带理论预言了很多拓扑非平庸的材料【1】,进而建立了拓扑材料数据库。然而,在拓扑平庸的材料库中,人们却忽略了一类奇特的“非常规”材料(Unconventional materials or Obstructed atomic limit)。
最近,中国科学院物理研究所/北京凝聚态物理国家研究中心王志俊特聘研究员、翁红明研究员和刘淼研究员指导博士生高嘉成,钱玉婷和贾华显,运用拓扑量子化学理论,通过高通量搜索找到423个非常规材料。这类材料的典型特征就是电子分布的轨道中心不在原子上,导致其表面、边、角上会存在比较自由的“活跃”电子态【2】。人们早期认知的共价键化合物属于这一类;我们还发现这些非常规材料在热电,储氢,催化,电子盐等特殊功能材料方向已经有了广泛的研究,这些特殊性质与其非常规的电子分布密切相关。

图1: 化合物AB的能带图示和高通量搜索流程图。(a)常规材料(原子轨道绝缘体),图示说明占据态能带对应的电子在实空间处在b威科夫(Wyckoff)位置上 (b)非常规材料(包含离域电子),图示说明占据态能带对应的电子在实空间处在b和c威科夫位置上。A 和B 原子分布在a和b威科夫位置,c位置为空。(c)高通量搜索这类非常规材料的流程图。
所谓常规材料是电子占据原子的局域轨道(atomic limit),也就是在形成化合物的过程中,电子可以由一个原子转移到另外一个原子,但始终被原子核紧密束缚。我们定义的非常规材料,其电子占据的实空间轨道中心在空位上(离域电子),不受(单一)原子核束缚,图一(a,b)给出了常规材料和非常规材料能带的示意图。对于一个具体材料的能带,利用拓扑量子化学的能带表示分解,我们可以得到这些能带对应的实空间轨道特征,包括电荷中心和轨道对称性,因此这种方法非常适合系统地挖掘非常规材料。为此我们设计如图一(c)所示的高通量搜索流程:1)通过第一性原理软件计算少数几个高对称k点的波函数;2)通过自主开发的能带表示计算软件IRVSP得到能带波函数在相关k点的小群表示;3)除了基本能带表示(eBRs)之外,我们还需要通过晶体中原子位置和价电子确定原子轨道能带表示(atomic band representations; aBRs),该步可以由开源程序pos2aBR完成。4) 通过对占据态能带作eBR分解和aBRs分解,筛选出非常规材料,即找出能带是eBR的组合,但不是aBR的组合的材料。5)进一步认证必要的(基本)能带表示(ρ@q),以确定离域电子的轨道特征。它的必要性由空位上的实空间不变量(Real-space invariants)来描述。
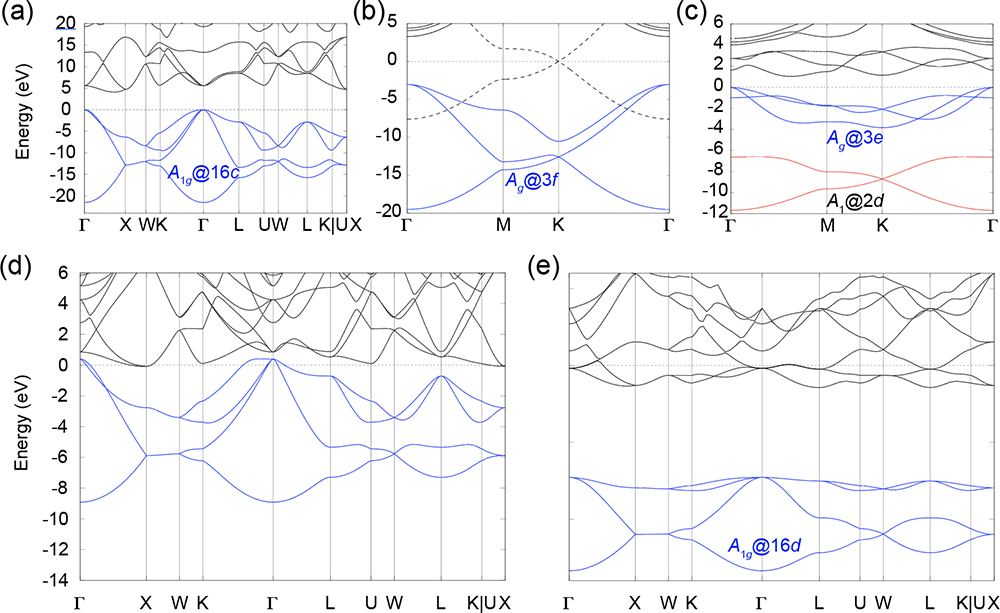
图二:(a)金刚石的能带以及对应的能带表示,图中蓝色占据态能带对应的电子态在实空间处在16c位置(空位);(b)石墨烯的能带及对应能带表示,(c)β-Sb的能带及能带表示;(d)和(e)分别是LiAl和LiAlH的能带和表示对比,其中氢原子占16d位置,在加入氢原子后,LiAl的占据态能带A1g@16d整体下移大约8 eV。
图二给出了一些非常规材料的能带结构以及占据态能带表示分解结果。共价化合物金刚石的成键电子在原子间化学键中心,因此在定义上也属于非常规材料,而二维的非常规绝缘体又可以被定义为二阶拓扑材料(在体系具有手征对称性的情况下)。此外,在搜索结果中还发现了许多奇特的功能材料,如由金属原子形成的半导体(Be5Pt 和 Na-hP4)、热电材料、价态不匹配的电子盐化合物、固体储氢材料等,这些功能材料通常具有以下特点:1)当空位上的电子暴露在界面上时会形成界面态;2)电子在空位上受到原子核的束缚小,对应电子功函数比较小;3)由于界面离域电子很容易与带正电的质子(H+)结合,有利于析氢反应的发生;4)由于离域电子的“活性“,这类材料在催化、电极化,以及超导性质方面都有一定的贡献。最近研究人员利用非常规材料Nb3Br8提出了一种无需外场的约瑟夫森二极管,研究结果发表在Nature 604, 653-656 (2022)。
对称性表示方法既有其高效性又有其局限性: 高效性体现在计算分析只需要少数几个高对称k点波函数的小群表示信息;局限性则是对称性表示相关理论所共有的局限。像对称性指标一样,对称性指标非零是拓扑材料的充分不必要条件,同样,占据态不是原子能带表示的组合也是非常规材料的充分不必要条件。但是,我们始终可以用Wilson loop的方法来直接计算能带对应的电荷中心,尽管比较费时。本研究组在近年来的研究中,已经将拓扑量子化学在磁群方向进一步扩展。目前拓扑材料的搜索适用于全部1651磁空间群[arXiv:2204.10556 (2022)]。为更好地将拓扑量子化学服务于第一性原理计算,研究组开发了一系列计算软件包[http://tm.iphy.ac.cn—资源共享]:计算第一性电子能带的表示的开源软件包IRVSP【3】;获得晶体结构对应的原子能带表示的开源程序包pos2aBR【4】;完成能带表示分解的线上代码eBR/aBR decomposition【2】; 实现原子结构到磁结构转变的代码pos2msg,搜索(磁性)拓扑材料,解析相容性关系和计算对称性指标的在线交互平台TopMat。
该项研究为探索具有应用前景的各类功能材料提供了新的思路。相关成果以“Unconventional Materials: the mismatch between electronic charge centers and atomic positions”为标题已在线发表在《Science Bulletin》上【2】。该工作获得了国家自然科学基金委、科技部、北京市自然科学基金委、中国科学院等项目的资助。